Overviews of concepts
eQTLs
An eQTL(expression Quantitative Trait Loci) is a region that is correlated with expression of a particular gene(transcript cluster) or probe set. This can either be in the same region as the gene, which is a cis-eQTL or it can be much further away on the same chromosome or a separate chromosome, which is a trans-eQTL.
For the recombinant inbred panels the eQTL is calculated for the gene/probe set across the RI strains. This results in a region between markers with the same calculated p-values being defined as an eQTL for the gene. In some instances no markers were close enough to a significant marker so the region is just the single marker, but this indicates we can't define a more exact region not that the SNP is responsible for the eQTL. There may be multiple locations, in most instances throughout the site you have the option to filter based on the p-value so only the most significant location(s) is shown. You can view the eQTLs for a specific gene through both the Gene and Region Detailed Transcription Information views. The gene view will allow you to view just the location of eQTLs for the gene. The region view will show the most significant eQTL in each tissue, for which data is available, or you may view a circos plot of all the eQTLs below a user set threshold.
Back to Top
Back to Top
bQTLs
Behavoiral Quantitative Trait Loci are regions that correlate to a continuous phenotype. A bQTL indicates a region that contains a genetic element that may contribute to the phenotype and allows you to narrow your search for the gene(s) responsible for a given trait.
Many of these have been published and can be explored for a region of interest in the Detailed Transcription Information section. If you have data for strains, which we have array data for you may create a login and import the phenotype data and calculate bQTLs and use the Detailed Transcription Information tool to explore expression and control of expression for genes in the region.
Back to Top
Back to Top
Heritability
Heritability indicates at either the probe set or transcript cluster (Gene) level which of these have a high genetic heritability.
The lower the heritability value the more environmental influence on expression is high compared to the
strict genetic influence. This allows sorting and filtering based on heritability to look for probe sets and transcript clusters
with high heritability and thus are more likely controlled by a genetic component, that may reside in an eQTL
region which you may explore further in the Detailed Transcription Information section.
How is heritability calculated?
The broad sense heritability is calculated for each probe set/transcript clusters separately using an ANOVA model.
For analyses on the Affymetrix Mouse 430 version 2 array, the broad sense heritability has been calculated on the public inbred mouse panel (20 strains) normalized using RMA with poor quality probes eliminated prior to normalization and the public BXD recombinant inbred mouse panel (32 strains) normalized using RMA with poor quality probe eliminated prior to normalization.
For analyses on the Affymetrix Mouse Exon array, the broad sense heritability has been calculated on the core transcript clusters from the public LXS brain recombinant inbred mouse panel normalized using RMA with poor quality probes eliminated prior to normalization.
For analyses on the Affymetrix Rat Exon array, the user must choose the tissue of interest (brain, heart, liver, or brown adipose tissue). The broad sense heritability has been calculated separately for each tissue on the core transcript clusters from the public HXB/BXH recombinant inbred rat panel normalized using RMA with poor quality probes eliminated prior to normalization.
Back to Top
How is heritability calculated?
The broad sense heritability is calculated for each probe set/transcript clusters separately using an ANOVA model.
For analyses on the Affymetrix Mouse 430 version 2 array, the broad sense heritability has been calculated on the public inbred mouse panel (20 strains) normalized using RMA with poor quality probes eliminated prior to normalization and the public BXD recombinant inbred mouse panel (32 strains) normalized using RMA with poor quality probe eliminated prior to normalization.
For analyses on the Affymetrix Mouse Exon array, the broad sense heritability has been calculated on the core transcript clusters from the public LXS brain recombinant inbred mouse panel normalized using RMA with poor quality probes eliminated prior to normalization.
For analyses on the Affymetrix Rat Exon array, the user must choose the tissue of interest (brain, heart, liver, or brown adipose tissue). The broad sense heritability has been calculated separately for each tissue on the core transcript clusters from the public HXB/BXH recombinant inbred rat panel normalized using RMA with poor quality probes eliminated prior to normalization.
Back to Top
Recombinant Inbred Panel
|
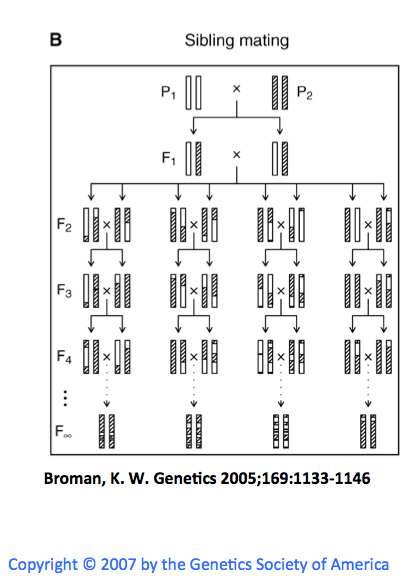
How are recombinant inbred panels generated?
The panels are started by crossing two inbred strains. Then crossing the siblings(F1) to create a population of heterozygous animals(F2). By continuously breeding(20+ generations) the siblings for each branch of the F2 generation you can get multiple strains of homozygous animals. This results in a panel of homozygous animals with different genomes(some combination of the parental strains) as a result of different recombination events in the previous generations. For many of the panels SNPs associated with each parent have been identified and each strain has been genotyped to provide information about what sections of their genome correspond to which parent. This information can then be correlated with phenotypes to provide insight into regions of the genome that contribute to a particular trait(behavioral(bQTLs) or expression of a gene or probe set (eQTLs). This is primarily what PhenoGen can be used to investigate using Arrays and for some organisms/datasets RNA-Sequencing.
What is the benefit?
These strains are available so phenotypic or behavioral data collected elsewhere may be correlated to data on PhenoGen. For example the BXH/HXB panels and ILSXISS panels have different alcohol consumption preferences. If you collected your own data on these strains you can import the mean and variance for each strain and calculate bQTLs, which would indicate regions of the genome correlated with a preference or aversion to alcohol consumption. You can also investigate regions that are correlated with expression of a gene or probe set, which can let you look for elements that control expression or even alternate splicing(when looking at control at the probe set level).
How can you use data from these panels on PhenoGen?
Phenogen includes a number of tools to let you take advantage of data collected on recombinant inbred panels.
Calculate regions associated with a phenotype (find regions/genes that may be responsible for a particular phenotype)
Calculate regions responsible for control of expression(find regions/genes/elements that may be responsible for expression at the gene or probe set level)
Analyze Expression (look at gene expression in different tissues and variance across multiple strains)
|
|
Additional Resources:
Jackson Lab-http://www.jax.org/smsr/ristrain.html
Wikipedia-http://en.wikipedia.org/wiki/Recombinant_inbred_strain
Pubmed-"Recombinant inbred strain panels: a tool for systems genetics", Curchill GA, Physiol Genomics. 2007 Oct 22;31(2):174-5.
Back to Top