
Working...Genes should load within 5-10 seconds. Regions depend on the size(ex. 10 Megabases may take ~1 minute).
Shortcuts:
{kind=link}
{kind=link}
{kind=link}
|
Region Image
|
View: |
Navigation Hints: Hold mouse over areas of the image for available actions.
Track Details
Region Summary
Track List:
Break down of track count*
*Note: Depending on the track settings some features may not be displayed and will not be reflected in the image above.
Features in Selected Track
Affy Exon Data Columns
The Affy Exon PhenoGen data displays data calculated from public datasets. Data is from the Public ILSXISS RI Mice.This dataset is available for analysis or downloading. To perform an analysis on PhenoGen go to Microarray Analysis Tools -> Analyze precompiled datasets. A free login is required, which allows you to save your progress and come back after lengthy processing steps.
Columns:
- Total number of non-masked probe sets
- Number with a heritability of >0.33 (Avg heritability for only probe sets >0.33)
- Number detected above background (DABG) (Avg % of samples DABG)
- Transcript Cluster ID corresponding to the gene with Annotation level
- Circos Plot to show all eQTLs across tissues.
- eQTL for the transcript cluster in each tissue
- minimum P-value and location
- total locations with a P-value < cut-off
Features Physically Located in Region Tab
This tab will display all the Ensembl features located in the chosen region.Data Summary:
- Gene Information(Ensembl ID, Gene Symbol, Location, description, # transcripts, links to databases)
- Probe Set detail (# Probe Sets, # whose expression is heritable(allows you to focus on expression differences controlled by genetics),# detected above background(DABG),(Avg % of samples DABG).
- Transcript Cluster Expression Quantitative Trait Loci (at the gene level)\ At the gene level, this indicates regions of the genome that are statistically associated with expression of the gene. The table displays the p-value and location with the minimum p-value for each tissue available. Click the view location plot link to view all of the locations across tissues.
Heritability
For each non-masked probe set on the Affymetrix Mouse Exon 1.0 ST Array, a broad-sense heritability was calculated using an ANOVA model and expression data from the ILSXISS panel. The heritability threshold of 0.33 was chosen arbitrarily to represent an expression estimate with at least modest heritability. Higher heritability indicates that expression of a probe set is influenced more by genetics than unknown environmental factors.Count indicates the number of probeset that have a heritability higher than 0.33.
The Avg is the average heritability among the probesets above 0.33.
Detection Above Background(DABG)
For each non-masked probe set on the Affymetrix Mouse Exon 1.0 ST Array and each sample, a Detection Above BackGround p-value was calculated for each probeset using Affymetrix Power Tools. A p-value less than 0.0001 was used as a threshold for detection. Using the p-value threshold of 0.0001, the proportion of samples from the ILSXISS panel that had expression values significantly different from background for a given probe set.Count is the number of probe sets for this gene that were detected above background in at least 1% of samples.
The Avg is the average across probe sets(only those probe sets >1%) of the proportion of samples that were above detection limits.
Affy Exon Data-eQTLs
The Affy Exon PhenoGen data displays data calculated from public datasets. Data is from the Public ILSXISS RI MiceThese datasets are available for analysis or downloading. To perform an analysis on PhenoGen go to Microarray Analysis Tools -> Analyze precompiled datasets. A free login is required, which allows you to save your progress and come back after lengthy processing steps.
Columns:
- Transcript Cluster ID- The unique Affymetrix-assigned id that corresponds to a gene.
- Annotation Level- Confidence in the transcript cluster annotation
- Circos Plot- Shows all eQTLs for a specific gene across tissues.
- eQTL for the transcript cluster in each tissue
- P-value from this region
- total other locations with a P-value < cut-off
Affy Exon Data-eQTLs
This tab requires the Java Plug-in to be installed and enabled. It is recommended to install the latest version of java for your computer. Note that only an older version is available on Mac OS X (Snow Leopard(10.6) and earlier), however, this version is still supported.On this tab you can explore data at the probe set level. The following views are available:
- expression levels of the parental strains for the inbred panel(ILS and ISS).
- heritability of each probe set.
- normalized probe set level expression across strains.
- exon-exon expression correlation.
This data is from the Public ILSXISS RI Mice dataset available under Microarray Analysis Tools -> Analyze precompiled datasets.
The data can be filtered to look at only probe sets that fall within a given transcript or based on detection above background or heritability, to help determine possible alternate splicing that may affect phenotypes.
Watch a quick navigation demonstration
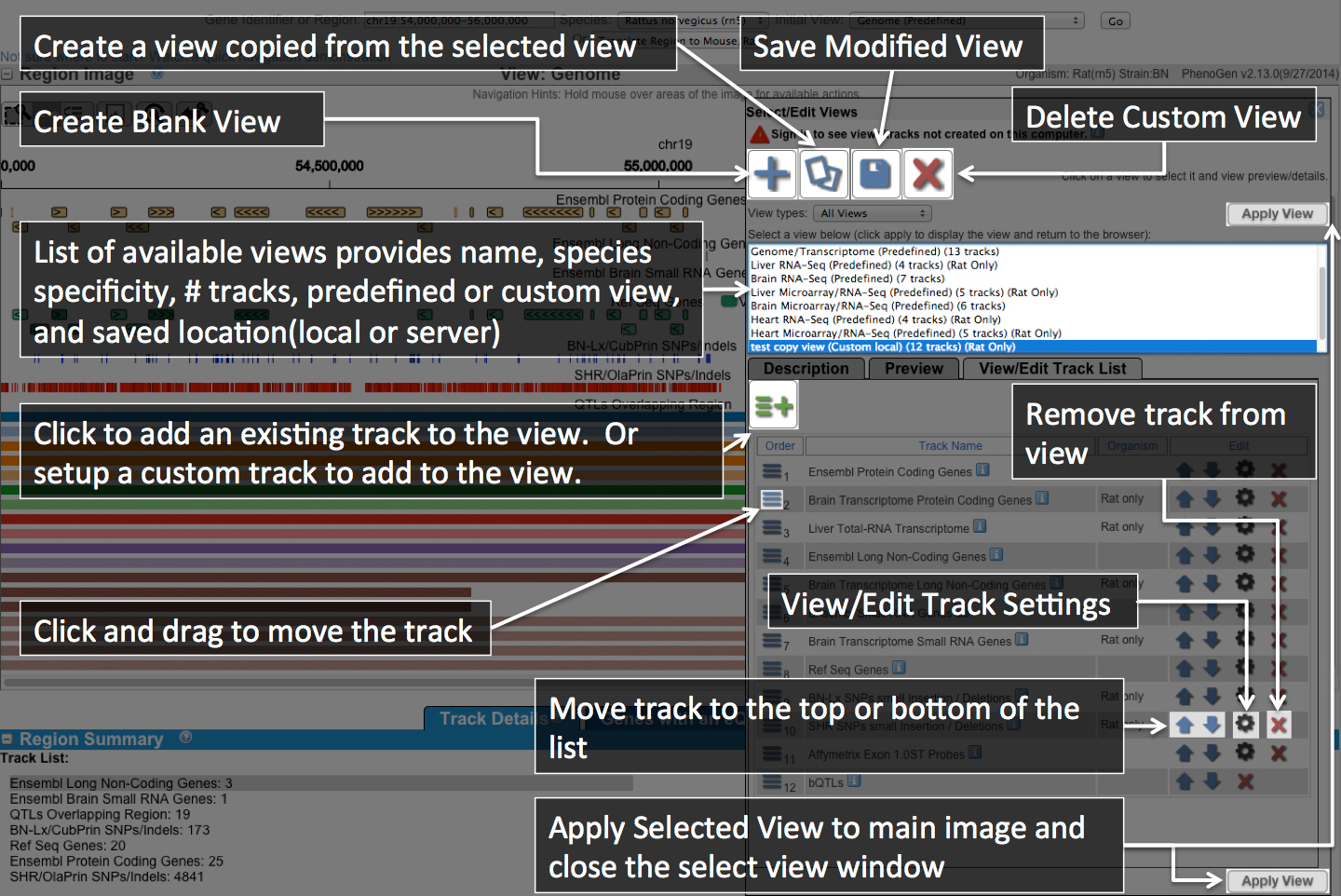
Note: This will overwrite the view you have saved for the Genome View.
Genome View
This view by default will show both Annotated Ensembl genes, along with bQTLs and SNPs/Indels. You may customize the tracks and views by clicking the Customize Image button. Changes to this view will be saved automatically if cookies are allowed so on the same computer you will get the same view when you select this tab again. You may always reset the current view to the default tracks/settings by clicking the Customize Image button and then the View Default Tracks button.Note: This will overwrite the view you have saved for the Genome View.
Watch a quick navigation demonstration
Note: This will overwrite the view you have saved for the Transcriptome View.
Transcriptome View
This view by default will show RNA-Seq reconstructed genes and Affymetrix Probesets. You may customize the tracks and views by clicking the Customize Image button. Changes to this view will be saved automatically if cookies are allowed so on the same computer you will get the same view when you select this tab. You may always reset the current view to the default tracks/settings by clicking the Customize Image button and then the View Default Tracks button.Note: This will overwrite the view you have saved for the Transcriptome View.
Watch a quick navigation demonstration
Note: This will overwrite the view you have saved for the Both View.
Both View (Genome and Transcriptome)
This view by default will show both Annotated and RNA-Seq reconstructed genes along with bQTLs, SNPs/Indels, and Affy Probesets. You may customize the tracks and views by clicking the Customize Image button. Changes to this view will be saved automatically if cookies are allowed so on the same computer you will get the same view when you select this tab. You may always reset the current view to the default tracks/settings by clicking the Customize Image button and then the View Default Tracks button.Note: This will overwrite the view you have saved for the Both View.
Watch a quick navigation demonstration
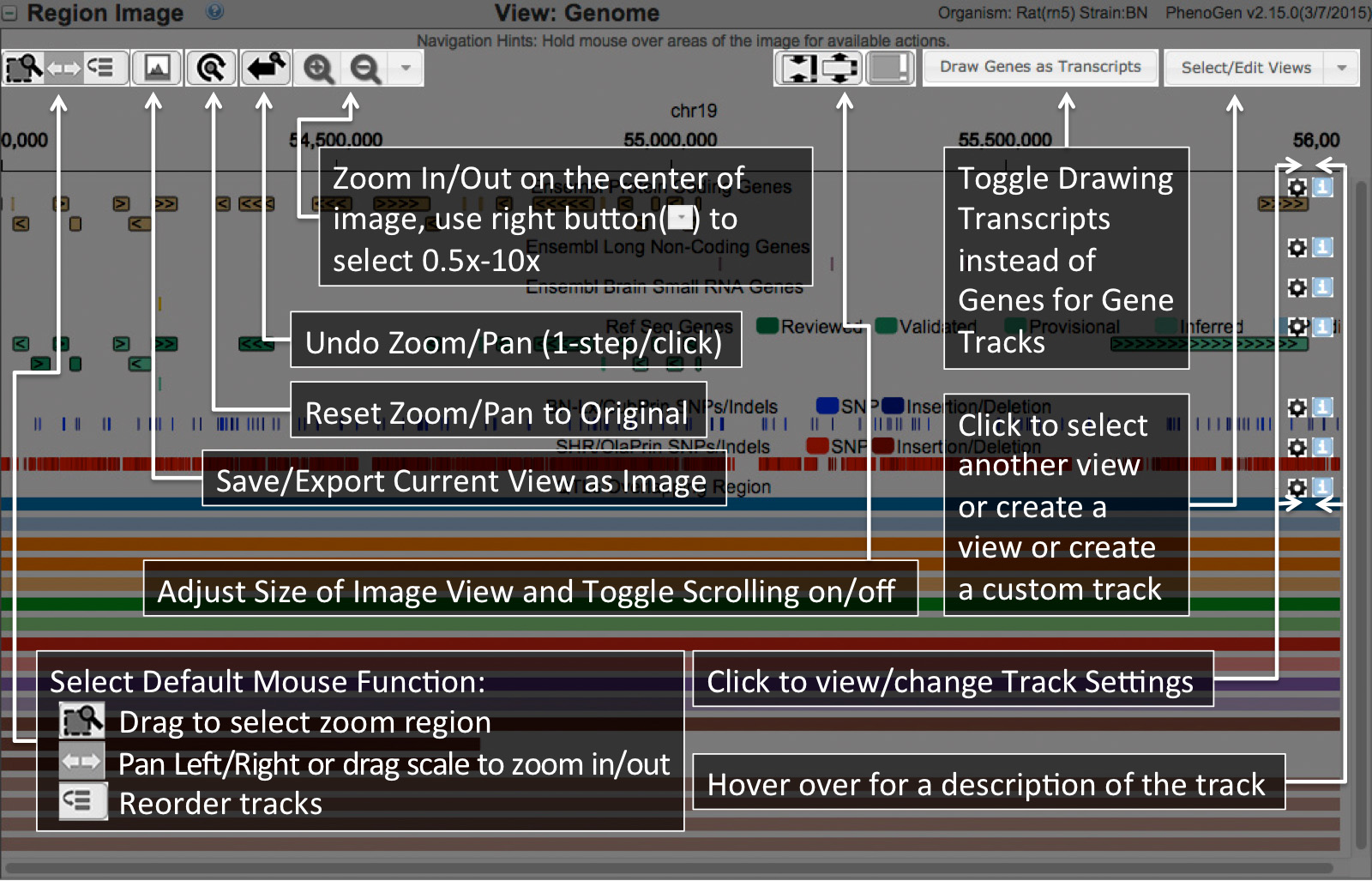
To see additional detail click on a feature. Features that are clickable will have a pointer (view example pointer Here) when you hold the mouse over them.
To Zoom in/out move the mouse over the scale at the top(view example pointer Here) of the image and drag right to zoom in and left to zoom out. Or you can double click on a feature to zoom in on it specifically.
To move left or right along the genome position your mouse over any track below the scale(view example pointer Here). You can click and drag to move in the direction you desire.
You may also move a track up or down to reorder the tracks to any order you wish. If cookies are enabled the tracks you select and order will be preserved between genes/regions/visits/etc for each seperate view(Genome/Transcriptome/Both).
To add/remove/change tracks in the image click the Customize Image button.
Region Image
The image below is generated by javascript which allows you to click on features to see additional detail, zoom in/out, and move along the genome.To see additional detail click on a feature. Features that are clickable will have a pointer (view example pointer Here) when you hold the mouse over them.
To Zoom in/out move the mouse over the scale at the top(view example pointer Here) of the image and drag right to zoom in and left to zoom out. Or you can double click on a feature to zoom in on it specifically.
To move left or right along the genome position your mouse over any track below the scale(view example pointer Here). You can click and drag to move in the direction you desire.
You may also move a track up or down to reorder the tracks to any order you wish. If cookies are enabled the tracks you select and order will be preserved between genes/regions/visits/etc for each seperate view(Genome/Transcriptome/Both).
To add/remove/change tracks in the image click the Customize Image button.
Region Summary
This section provides tables, charts, images sumarizing the features in the current region of the genome or genes with eQTLs in the region. You may switch between the two options using the blue tabs immediately above. For more detail see the help for each tab.You can show or hide this section at any time use the + or - icon to the left of the section header. This section is automatically hidden when a feature is selected in the image above. If you would like to view it again you may always expand the selection again with the + or - icons.
Track Details
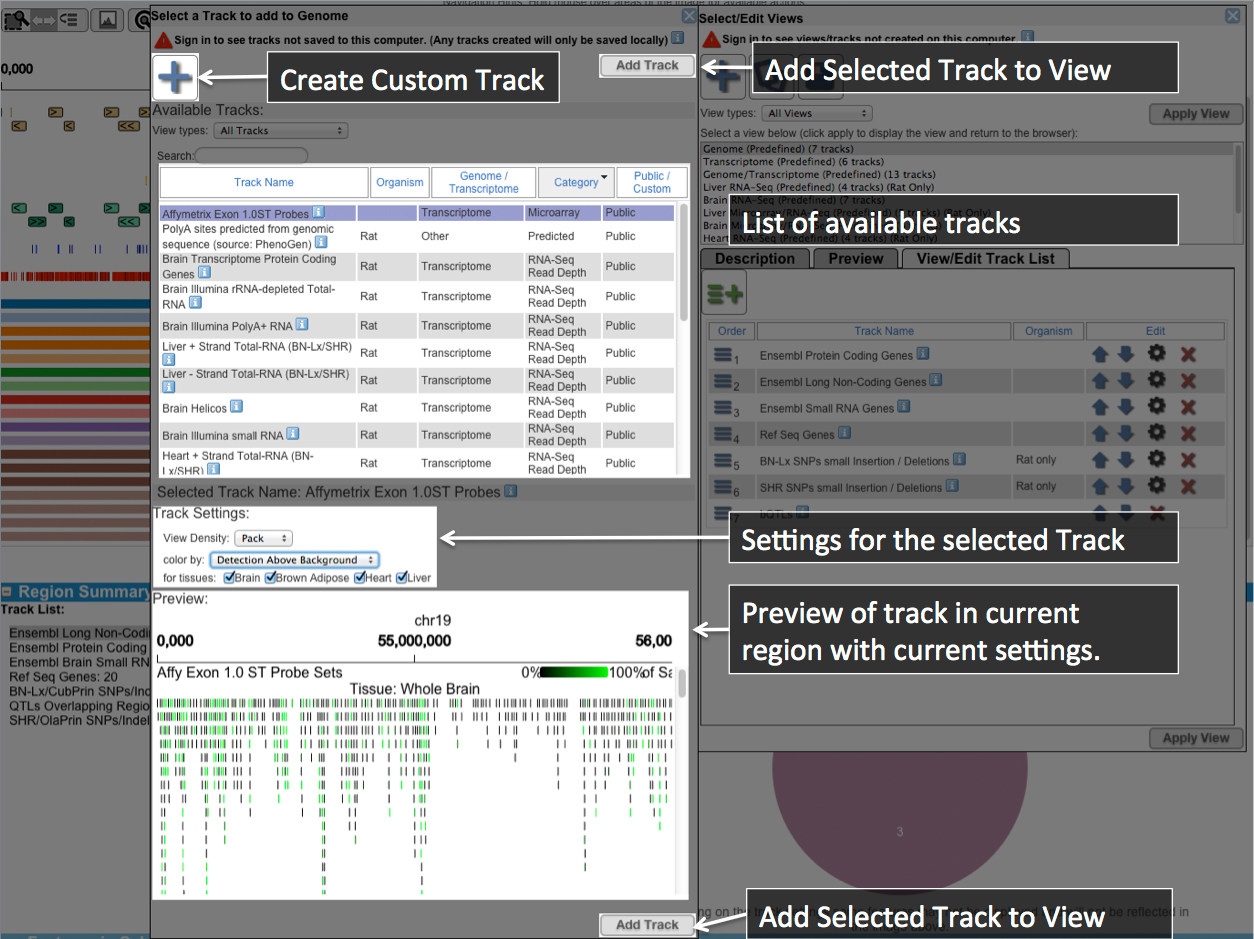
This section lists tracks that are available to summarize. This view provides a break down of features in a track for example if you are view annotated and reconstructed Protein Coding genes it will provide a breakdown of Ensembl annotated and unique to RNA-Seq genes or for SNPs/Indels will display the proportion of SNPs/Insertions/Deletions in a region for the selected track. Most of the tracks also can display a table listing each feature in a track. These tables are displayed for the selected track when the Track Feature Table is expanded.UCSC Image Controls
The controls in this section determine what information is included in the image above and tables below.Each track includes information about it. Hold your mouse over the
Check the box next to the track you would like to include in the image. The image should then be generated and appear shortly. For some tracks, you may control the way the track is displayed. You can change this by selecting the option next to the track.(ex. )
Dense-This is the most compressed view. Generally with no labels for features and all features compressed into a single line of the display.
Pack-Fits multiple features per line such that features and their labels do not overlap.
Full-Each entry in the track has a dedicated line of the display. The label for each entry can be found at the far left. Entries will be order from top to bottom in order of their occurrence from left to right.
The best option depends on what you are looking for so try them and find what will work for you.
eQTLs
eQTL are calculated for transcript clusters, which are approximately equivalent to a gene-level estimate of expression by combining multiple probesets that target the same gene. The combination of probesets for each transcript cluster is defined by Affymetrix to include probesets that target regions that are included in all transcripts of a gene.Columns:
Transcript Cluster ID- The unique ID assigned by Affymetrix.
Location-The chromosomes and base-pair coordinates where the gene is located.
Annotation level- Related to the confidence in the gene. Core is the highest confidence. This level tends to correspond very closely with the Ensembl gene annotation. Extended is lower confidence and may include additional regions outside of the Ensembl annotated exons. Full is even lower confidence and includes additional regions beyond the Ensembl annotations.
Genome Wide Associations- A way to view all the locations with a P-value below the cutoff selected. Circos is used to create a plot of each region in each tissue associated with expression of the gene selected.
Tissue Columns
These columns summarize the data for each tissue.
Total # of locations with a P-value < (Selected Cutoff)- The number of locations associated with expression below a cutoff selected in the Filter List section above the table.
Minimum P-Value Location- The P-value and location of the minimum P-Value for the given tissue. P-Value is in black, above the location in blue. Click the location to open a Detailed Transcription Information window for that location.
bQTL Tab
The bQTL tab allows you to view bQTLs that overlap with the region.What is a bQTL?(View detailed bQTL information) A Behavioral Quantitative Trait Loci or bQTL is a region that is associated with a particular phenotype or behavior (thus bQTL).
How is it calculated? bQTLs can be found for Recombinant Inbred Panels by measuring a trait/behavior across strains in the panel and then correlating the values to the genotype of each strain between markers. Based on that correlation regions can be found that are correlated with a particular phenotype. These are the regions listed in the table under this tab. They may indicate that a gene or other feature is somehow influencing the phenotype.
eQTL Tab
This tab shows the genes that might be controlled by a feature in the chosen region. There is at least an eQTL with a P-value below the cutoff in the highlighted(Blue) for the selected region in one or more tissues. However, the actual region may just overlap with the current region, so the eQTL region associated with the P-value may be a different than the current region.The Circos plot shows where the genes with eQTLs in this region are physically located.
The table below the Circos plot lists all the genes and eQTLs for each gene in each tissue. Use the View Location Plot link to view all the eQTLs for a gene in a Circos plot that shows each eQTL for the selected gene.
Finally, to view detailed transcript and probe set data, click on a Gene Symbol to display a summary specific to that gene.
What is an eQTL? (View detailed eQTL information) An eQTL is a region that is correlated across recombinant inbred strains to expression of a gene(or in the case of our Affy data displayed, Transcript Cluster) or probe set. This may indicate some feature in this region is influencing expression of the gene with a significant eQTL in the region.
How is it calculated? eQTLs can be found for Recombinant Inbred Panels by measuring expression across strains in the panel and then correlating the values to the genotype of each strain between markers. Based on that correlation, regions can be found that are correlated with expression. In the table, the region overlaps with one of the regions that is assigned a P-value below the cutoff you selected. This may indicate that a gene or other feature in this region, or one of the other regions with a significant P-value, is somehow influencing the expression of the gene.
What does an eQTL for a transcript cluster mean? At the transcript cluster level, this is an eQTL for a gene and not individual probe sets. For now, this is the only level available.
WGCNA Tab
Weighted Gene Co-expression Network Analysis provides information on transcripts that have correlated expression by grouping them into modules. This tab provides information on the transcripts and genes grouped together into a module. For a region, any module that contains a transcript from the region is displayed. You can select each module individually and view the correlations between transcripts and get additional information on transcripts and genes. You can also view eQTLs for the module, which indicates possible regions that may be correlated with regulation of transcripts in the module. In this view you can also see the genes in the region that may contribute to regulation.WGCNA data is based on the Affymetrix microarray data set either HXB/BXH Recombinant Inbred Panel(Rats) or ILS/ISS Recombinant Inbred Panel(Mice). However, grouping of probe sets into transcripts is based on the tissue specific RNA-Seq transcriptome reconstruction(accessible in a track of the genome/transcriptome browser) and correlation of microarray expression.
Future updates will also indicate miRNAs, transcription factors, and miRNAs validated and predicted to target genes in the module. For Rats future updates will provide heart and liver data as well.
Gene List - WGCNA Tab
Weighted Gene Co-expression Network Analysis provides information on transcripts that have correlated expression by grouping them into modules. This tab provides information on the transcripts and genes grouped together into a module. For a gene list, any module that contains a transcript from the gene list is displayed. You can select each module individually and view the correlations between transcripts and get additional information on transcripts and genes. You can also view eQTLs for the module, which indicates possible regions that may be correlated with regulation of transcripts in the module. In this view you can also see the genes in the region that may contribute to regulation.WGCNA data is based on the Affymetrix microarray data set either HXB/BXH Recombinant Inbred Panel(Rats) or ILS/ISS Recombinant Inbred Panel(Mice). However, grouping of probe sets into transcripts is based on the tissue specific RNA-Seq transcriptome reconstruction(accessible in a track of the genome/transcriptome browser) and correlation of microarray expression.
Future updates will also indicate miRNAs, transcription factors, and miRNAs validated and predicted to target genes in the module. For Rats future updates will provide heart and liver data as well.
eQTL Tab
This tab shows a Circos Plot of the eQTLs associated with the selected Transcript Cluster(Gene). The lines connect from the location of the gene to the region/SNP associated with expression of the gene. Each tissue contains yellow/black lines that indicate the P-value of that association. Yellow indicates the P-value is below the selected threshold. The controls available under this tab let you select the p-value, chromosomes, and tissues(if applicable) to include in the image.Circos Plot eQTL Gene Locations
This plot shows all of the genes that have an eQTL in the region entered. These genes correspond to the genes listed in the table below. If a higher number of genes are located in nearly the same region only the first 2-3 may be displayed.The plot can be hidden using the +/- button. The size of the plot can also be controlled using the Adjust Vertical Viewable Size drop down list.
When your mouse is inside the border below, you can zoom in/out on the plot. When your mouse is outside the border you can scroll normally. The controls inside the image can be used to zoom in and out and reset the image. You can also click-and-drag to reposition the image.
You can download a PDF of the image by clicking on the download icon(
DAVID
A list of genes can be imported into the DAVID web site for additional information about function and also to look for a significant enrichment in pathways, which might imply some biological meaning. The link, when available, will open the summary page where you can explore the different DAVID tools.Currently only lists of 300 genes are less are supported. This is a limit of the method used to submit genes to the DAVID web site. We will either be replacing the site or supporting a different method to allow longer lists. If you perform filtering to get the list below 300 you will be able to click a link and immediately analyze data on the site, Otherwise you can copy one of the ID columns such as Gene IDs(Ensembl IDs) and submit them on your own.
Gene Detail Tab
The Gene Detail Tab provides summary information and links related to the selected gene. There are three sections on this page. General information includes location, gene symbol, links to other databases, Ensembl transcripts, and RNA-Seq reconstructed transcripts if available.The Probe Set Information section provides a summary of probe sets across all available tissues including expression above background and heritability. The Probe Set Level Data tab has more detailed information allowing you to look at each probe set.
The eQTL section provides information on the location of the minimum P-value for each available tissue. A more complete list is provided on the eQTL Tab.
Gene eQTL Tab
The eQTL Tab provides expression Quantitative Trait Loci for a selected Affymetrix transcript cluster representing the gene selected. It provides all the loci with a P-value below a selected cut-off. You can change the cut-off and include/exclude chromosomes as desired. If a gene has multiple transcript clusters assigned then they all will be available. The highest confidence transcript cluster is selected first, for example if there was a core and a full transcript cluster the default is the core transcript cluster.Gene Probe Set Details Tab
The Probe Set Detail Tab allows you to view detailed probe set level data. This includes RI panel parental differential expression, detection above background, heritability, expression, and probe set correlation. You can view the data across panel strains and tissues where available.This runs as a signed Java applet in a new window, so you need an updated JRE and must approve it to run and may require allowing the popup. It will prompt you to install the latest JRE and will prompt you to make sure you want to run it. There are some errors with specific JREs, browsers, and operating system combinations. You should be notified if we have discovered a problem with your combination of browser, OS and JRE, but please try a different browser or computer if you run into issues. If you let us know through the feedback form we will try to find a solution.
miRNA Targeting This Gene Tab
The miRNA Targeting Gene (MultiMiR) Tab displays a summary of miRNAs that are predicted or validated to target a particular gene. You can view detailed information on the miRNA and get a list of everything it targets or is predicted to target.miRNA Targets Tab
The Genes Targeted by this miRNA (multiMiR) Tab displays a summary of genes that are predicted or validated as targets of the selected miRNA. You can view detailed information on the miRNA or genes and get a list of miRNAs that target a gene from the list.Gene WGCNA Tab
Weighted Gene Co-expression Network Analysis provides information on transcripts that have correlated expression by grouping them into modules. This tab provides information on the transcripts and genes grouped together into a module. For a selected gene, any module that contains a transcript of the selected gene is displayed. You can select each module individually and view the correlations between transcripts and get additional information on transcripts and genes. You can also view eQTLs for the module, which indicates possible regions that may be correlated with regulation of transcripts in the module. In this view you can also see the genes in the region that may contribute to regulation.WGCNA data is based on the Affymetrix microarray data set either HXB/BXH Recombinant Inbred Panel(Rats) or ILS/ISS Recombinant Inbred Panel(Mice). However, grouping of probe sets into transcripts is based on the tissue specific RNA-Seq transcriptome reconstruction(accessible in a track of the genome/transcriptome browser) and correlation of microarray expression.
Future updates will also indicate miRNAs, transcription factors, and miRNAs validated and predicted to target genes in the module. For Rats future updates will provide heart and liver data as well.
WGCNA Views
There are currently five views available the module list, the module, the eQTL, the Gene Ontology, and the miRNA(MultiMiR) views. Above both of the individual views is a list of modules. Following is a description of each view.Module List
Initially you will be given a list of all modules that correspond to your source(region, gene, or gene list). This list includes any module containing a transcript from your source. The modules are ordered by the number of transcripts in the module denoted by the number in the middle of the module. They are sized relative to each other based on the total number of transcripts in the module. To allow you to start viewing data more quickly modules will be displayed as they download which is why the module list may change while the loading image is displayed if viewing a large number of modules. You can view additional details by holding your mouse over a module and clicking on a module will select it and bring up one of the following views based on your selected view.Module View
The module view displays up to 150 transcripts within a module. Modules larger than that will have a warning icon indicating the graphics have cutoff the remaining transcripts. The 10-20 largest modules in each dataset will be truncated. The table below the graphics will display all of the transcripts even when the graphics do not.Each node corresponds to a transcript, which is labeled with its corresponding gene symbol if available. Nodes are order from the top center clockwise back to the top center. Nodes are ordered by taking the sum of absolute value of all correlation links for a node. This value is also used to size each node. Thus the most connected node(Hub Gene) is the first node in the image. The links between nodes are colored based on the direction of the correlation. The default is green for a positive correlation and red for a negative correlation. A control above the image allows for selecting of blue/yellow or the opposite red/green coloration. Links are sized based on the absolute value of the correlation.
You can use the sliders above the image to change the minimum and maximum cutoffs for the links displayed. You also have the option to hide all links until you hover over a node at which point only links to that node are displayed.
eQTL View
The eQTL view displays the calculated eQTLs along the genome for the module. The yellow bars along the genome denote locations with a ‐log(P-value) above the selected cutoff. Links are also drawn from the module(center) to the locations above the cutoff. The highest P-value is highlighted with a yellow link.The outer section of the image shows the genes and locations of genes for each gene with transcripts in the module. This allows you to quickly look at overlap of eQTLs with genes that may contribute to regulation.
Gene Ontology View
The GO term view summarizes the GO terms assigned to all of the genes within the module that have GO terms assigned. It does not reflect every gene and the number of genes may vary from GO domain to GO domain within a module. The GO term annotations are from the current version of Ensembl. The image is arranged in a sunburst plot where the GO domain term is at the center and children terms are displayed at successively lower levels moving out. The number of levels is initially limited. You can use the controls above to change the root GO domain or increase/decrease the levels displayed. You also can navigate through the tree by clicking on a term to make it the root of the image and display x number of levels below the newly selected term. The table below reflects this tree structure and allows you to expand it. If you select a new root in the image the root of the table changes as well and if you click on a row of the table that becomes the root of the table and image.miRNA (multiMiR) View
The miRNA view is the same as the module view with the miRNAs displayed in the center. miRNAs are displayed with different sizes and colors in the center of the image. The size is based on the number of genes targeted in the module. The color of nodes varies to make finding an miRNA easier, but is not tied to data. When the mouse is over a miRNA, links are drawn to targeted genes. Yellow indicates a validated target. Blue indicates a predicted target. If the option is selected green and red links will be displayed between targeted genes showing correlation between target genes or optionally all correlations from a targeted gene. The width of the lines indicates the level of support or correlation for links between miRNAs and Genes or Genes and other Genes respectively. When the mouse is over a gene the miRNAs are filtered to only show miRNAs that target that gene. The links displayed are the same as links displayed when holding the mouse over a miRNA depending on the display options selected.For transcripts around the edge of the image up to 150 transcripts within a module will be displayed. Modules larger than that will have a warning icon indicating the graphics have cutoff the remaining transcripts. The 10-20 largest modules in each dataset will be truncated. The table below the graphics will display all of the transcripts even when the graphics do not. Each node corresponds to a transcript, which is labeled with its corresponding gene symbol if available. Nodes are order from the top center clockwise back to the top center. Nodes are ordered by taking the sum of absolute value of all correlation links for a node. This value is also used to size each node. Thus the most connected node(Hub Gene) is the first node in the image. The links between nodes are colored based on the direction of the correlation. The default is green for a positive correlation and red for a negative correlation. A control above the image allows for selecting of blue/yellow or the opposite red/green coloration. Links are sized based on the absolute value of the correlation.
The table below summarizes either the miRNAs that target genes in the module. It can be grouped by miRNA, effectively displaying a list of all miRNAs with the option to see the genes each targets or by gene displaying a list of each gene and the miRNAs that target that gene. The current can be exported by using the Export CSV button.